
近日,中科院武汉植物园李治中和陈进明课题组联合永利集团3044在权威期刊Molecular Ecology Resources(IF=7.09)上发表了题为“The chromosome- level genome of a free- floating aquatic weed Pistia stratiotes provides insights into its rapid invasion”的研究论文,该研究通过PacBio和Hi-C测序构建了高质量的大薸基因组,随后通过比较基因组分析初步解析了大薸的进化与快速入侵的分子机制,并在大薸基因组中鉴定到了85个抗病相关基因家族(NBS-LRR),从而为大薸快速入侵机制的研究提供了新见解。菲沙承担了本研究中基因组的测序与Hi-C分析工作。

图1 文章发表信息
研究背景
大薸(Pistia stratiotes)隶属于天南星科,是一种自由漂浮的植物,广泛分布于热带和亚热带地区。由于繁殖速度快、形态结构独特、生态危害性强,大薸被列为最具入侵潜能的物种之一,其对入侵地区的经济和自然生态系统造成了严重破坏。
近年来,全基因组测序已成为研究物种适应的重要工具,并已成功应用于多种入侵物种的生态适应性研究,例如薇甘菊、凤眼莲、空心莲子草等。但目前对于水生入侵植物的基因组学研究较少,其快速入侵的遗传机制在很大程度上未被探索。此外,虽然已有研究表明大薸抗病基因家族的扩张有助于其环境防御与广泛分布,但尚不清楚这些基因是否与大薸对新环境的快速适应相关。基于此,通过构建大薸高质量的参考基因组,可为其快速入侵机制的研究提供新见解。
研究思路
材料:大薸(Pistia stratiotes)
测序策略:PacBio+Hi-C
研究结果
1
// 大薸基因组的组装与注释
通过Survey分析,研究者发现大薸基因组的杂合度高达2.16%,随后进行了高深度的PacBio(531×)和Hi-C测序。通过组装,获得的大薸基因组大小为311.87Mb,contig N50=1.08Mb,并将96.03%的序列锚定到14条染色体上。BUSCO评估基因组完整性为86.9%,这说明研究者获得了高质量的大薸基因组。
通过注释,研究者获得了20356个蛋白编码基因,其平均长度为225.39bp,BUSCO评估注释完整性为85.3%;大薸基因组中的重复序列占比为54.47%,其中18.54%的为LTR逆转录转座子。此外,研究者还预测得到89个miRNA、164个rRNA、306个tRNA、213个sRNA。
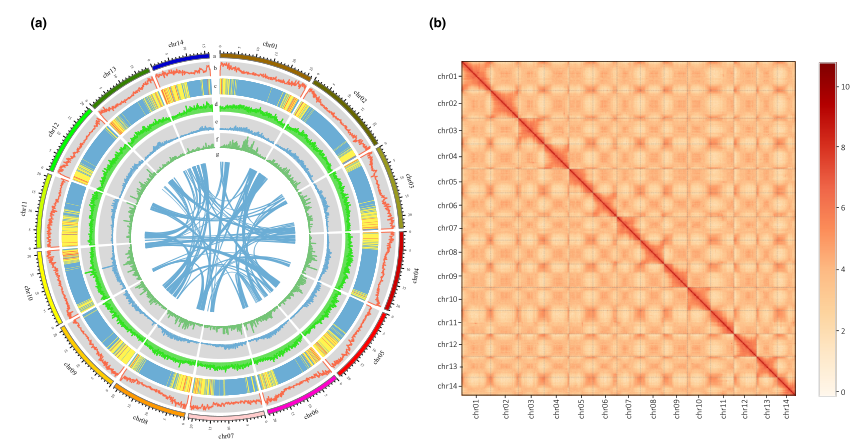
图2 大薸基因组特征
2
// 大薸基因组的进化及WGD事件分析
通过选取16个物种进行比较基因组学分析,研究者共获得了3175个单拷贝同源基因。系统进化分析表明,大薸与天南星科的C. esculenta亲缘关系较近,两者在60.78个百万年前产生分化。基因家族扩张收缩分析表明,大薸基因组中共有485个基因显著扩张,显著扩张的基因主要富集在与鞘脂代谢”、“植物-病原体相互作用”和“生物素代谢等生物合成相关的通路中,这些结果说明了扩张基因增强了大薸的抗性与适应性。此外,进一步将大薸与其它4个物种进行近家族聚类分析,发现大薸特有的基因家族为266个,主要富集在与“植物-病原相互作用”、“类黄酮生物合成”和“植物昼夜节律”相关的通路中,而缺失的基因家族主要与大薸的生活方式相关。
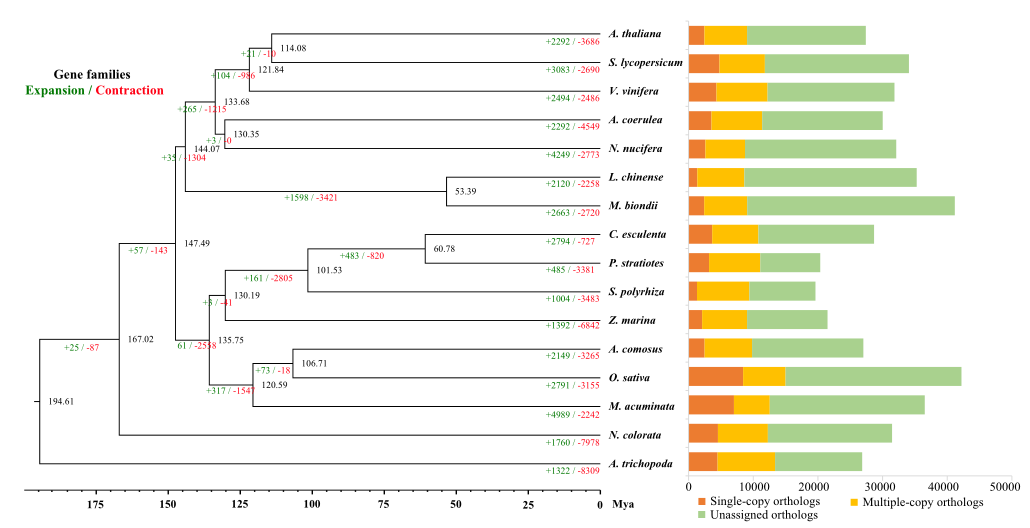
图3 大薸与近缘种的系统进化分析
通过共线性分析,研究者发现大薸与水稻的共线性深度比为2:4,这预示大薸可能经历了两次WGD事件,4DTv和Ks分析也确认了大薸的两次WGD事件,且这两次WGD事件大薸与近缘物种共享。对WGDs后的重复基因进行GO和KEGG分析,其中大部分涉及基因表达、代谢途径和信号转导途径的调控,这说明WGD事件为大薸基因组进化中的抗逆性和适应性提供了遗传基础。
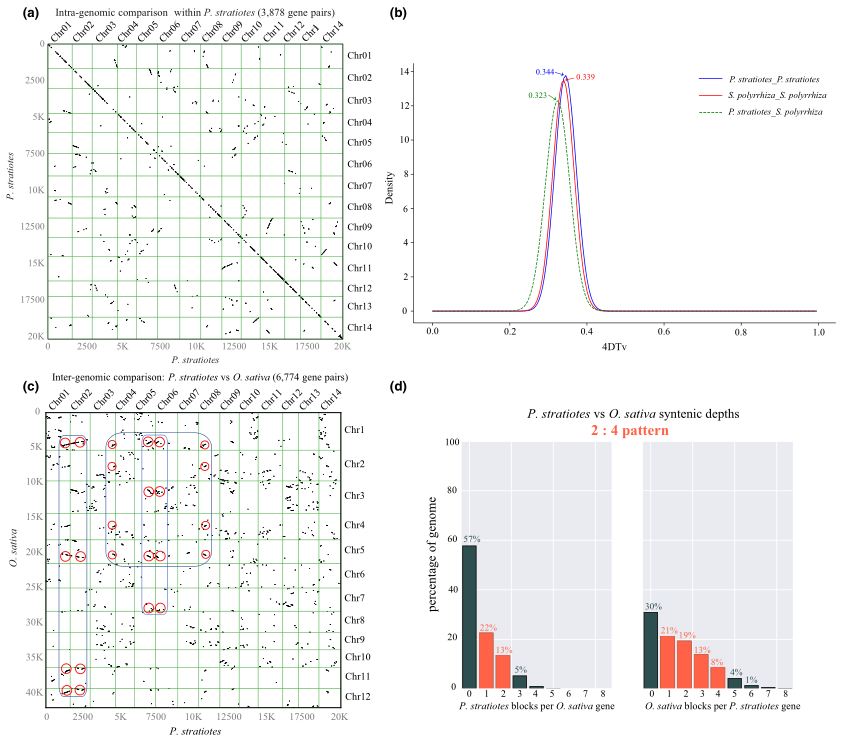
图4 大薸的WGD事件分析
3
// 大薸中抗病相关基因家族的鉴定
植物利用具有NBS-LRR结构域的细胞内蛋白质去抵抗病原体,研究者在大薸中鉴定到85个NBS-LRR基因,包括84个CNL和1个RNL。大多数NBS-LRR基因在染色体上呈簇状分布,其中大多数分布在6号染色体上。此外,大薸中NBS-LRR主要发生串联复制事件,且正在经历纯化选择。
最后,研究者将NBS-LRR基因家族的系统发育树与真实物种树进行了比较(分为三类:CNL、TNL、RNL),发现大薸中NBS-LRR发生了实质性的扩张,这可能与其发生的串联重复事件相关。此外,大薸中的RNL基因和拟南芥中的6个RNL基因聚在一起,这与大薸的抗性可能相关。
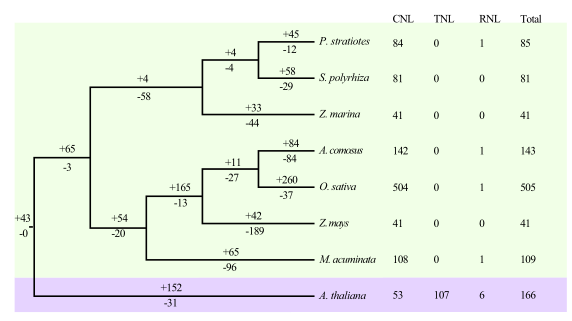
图5 八种植物NBS-LRR基因的进化树分析
 微信公众号
微信公众号


 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|
