
我们知道,传统的二代转录组建库需要对反转录的cDNA进行打断,无法直接获取单个RNA分子5ˊ到3ˊ的全部序列。而基于PacBio SMRT平台的全长转录组测序,无需打断,能够直接读取反转录的全长cDNA序列。字面意义来看,三代全长转录组建库不对cDNA进行打断,但事实上,做过三代全长转录组测序的小伙伴都明白全长转录组建库对cDNA片段筛选却是有套路的。
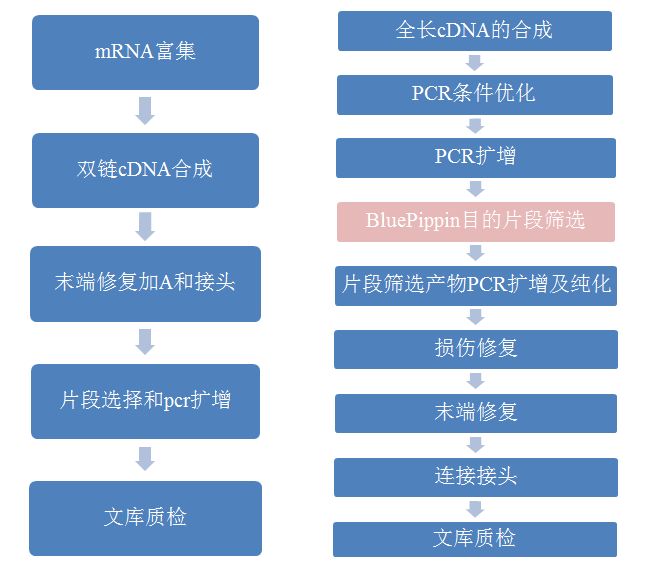
二代转录组及全长转录组(ISO-Seq)建库流程图
PacBio平台测序原理决定其构建分片段文库。由于mRNA长度不同,cDNA文库小分子(短片段)在落入零模波导孔时会被优先选择loading,大分子(长片段)可能存在loading遗漏。为了获得长片段序列信息,PacBio测序构建文库时要根据物种的mRNA长度进行不同片段大小的文库区分,分段文库越多,得到的全长转录本越全面。
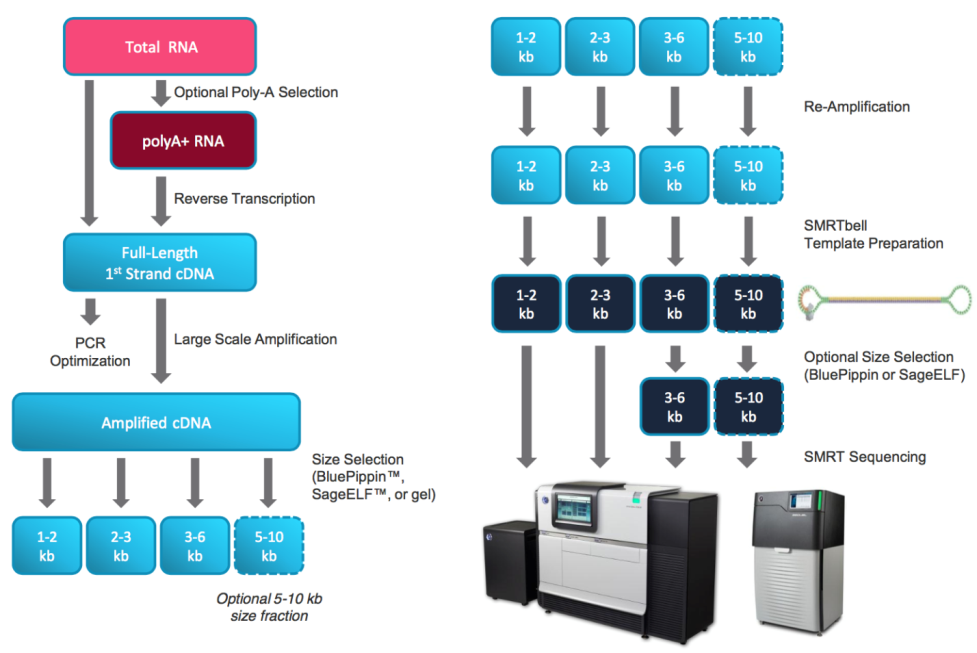
全长转录组测序实验流程图
最初PacBio RSⅡ应用时,全长转录组测序一般推荐至少构建三种文库类型,1-2Kb、2-3Kb和>3Kb文库。但随着Sequel Binding Kit 3.0、Sequel Sequencing Kit 3.0与Sequel SMRT Cell 1M v3测序升级以来,片段分选已不再是主流建库策略,1M v3芯片的高数据量产出基本可满足较长转录本检出。复杂物种同时想要获得更为全面的转录本或者检出稀有转录本,此时会针对性的对大片段进行分选建库(>5Kb or >6kb),并依据物种复杂程度、基因大小和研究目的来确定合适的测序数据量,测序数据量越多,检测到的全长转录本越全面。
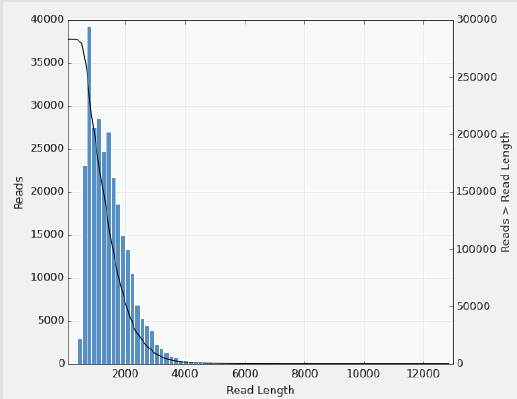
FLNC长度分布(1M v3芯片)
由于转录组信息呈动态变化且存在空间和时间差异,某一组织得到的全长转录本对该物种其它组织部位、某一物种特定发育时期的全长转录本对其它发育时期可能不很全面或不太适用。
为打破这种局限,目前已有的全长转录组建库通常使用混样建库。要想获取某一物种相对全面的转录本信息,需对该物种的不同部位进行取样,提取得到的totalRNA等量混合[2]。获得某个特定组织部位的转录本信息,则是对该组织的不同发育时期进行混样;若想要研究某种胁迫处理对物种的影响,需要分别对照组和处理组进行全长转录组测序(两个全长)对比分析[3]。
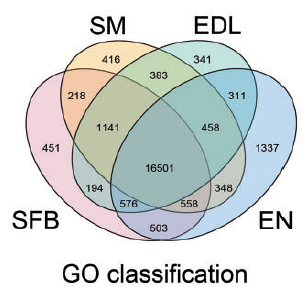
不同组织isoform的GO注释
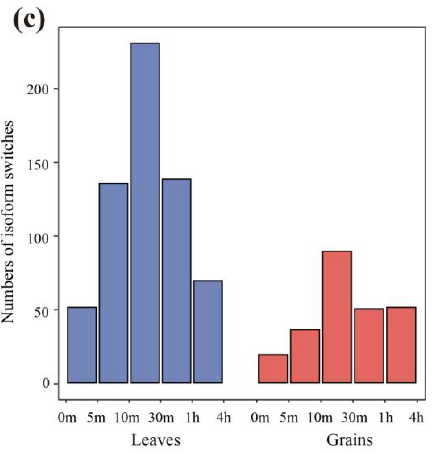
非生物胁迫条件下不同发育时期组织isoform的数量
在混样策略不变的前提下,随着更高数据产出的PacBio Sequel Ⅱ 8m芯片的面世,个性化大片段文库分选构建也将变得不再必要。这将大大缩减建库周期,明显降低测序成本。
应广大科研老师的需求,小编在文章的最后为您附上最详尽的三代全长转录组建库流程,供参考:
1、全长cDNA的合成
使用Clontech的SMARTer™ PCR cDNA Synthesis Kit合成mRNA的全长cDNA。
2、PCR条件优化
PCR扩增富集合成的cDNA,通过循环优化确定PCR的最佳条件。
3、PCR扩增及纯化
选取最适的PCR反应条件,进行cDNA一链产物的PCR扩增;PCR产物纯化。
4、Blue Pippin筛选目的片段
取部分cDNA利用BluePippin进行片段筛选,Blue Pippin切胶产物纯化
5、片段筛选产物PCR扩增及纯化
将筛选片段进行大规模PCR并进行产物纯化,以获得足够的cDNA总量。
6、DNA损伤修复、末端修复、连接接头、核酸外切酶消化及纯化
对全长cDNA进行末端修复,连接SMRT哑铃型接头,进行核酸外切酶消化,去除cDNA两端未连接接头的序列及消化产物纯化,获得上机文库。
7、文库质检
取适量文库进行Qubit定量,计算文库浓度;取适量文库进行 2100检测,确定文库大小。
参考文献
[1] Dong L,etal.Singlemoleculerealtime transcript sequencing facilitates common wheat genome annotation and grain transcriptome research [J]. Bmc Genomics, 2015, 16(1):1-13.
[2] Li Y, et al. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing [J]. DNA Research, 2018, 25(4), 421–437.
[3] Wang X M, et al. Hybrid sequencing reveals insight into heat sensing and signaling of bread wheat [J]. The Plant Journal, 2019.
 微信公众号
微信公众号


 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|
