
茶,起源于中国,发扬于全世界,是中华民族的举国之饮。自唐陆羽《茶经》“所庇者屋屋精极,所着者衣衣精极”至陈椽教授耗毕生精力著《茶业通史》,茶及其所形成的“茶文化”便亘古流长。时至今日,依工艺和品质差异,茶概括分为六类:红茶、绿茶、乌龙茶、黄茶、黑茶、白茶。红茶浓鲜,绿茶鲜爽,乌龙岩韵,黄茶甜爽,黑茶陈醇,白茶清甜,众多口感的香茶,除让人味觉大呼过瘾外,其背后的分子与代谢机制也让科研人员倍感着迷。

图1 六大名茶概述图
随着多组学尤其是基因组学的快速发展,科研人员在“六大名茶”的遗传育种、起源进化、结构变异、代谢物合成解析等方面取得重大突破,研究成果相继发表在PNAS、MP、NC等著名期刊上。值此机会,小编将“六大名茶”基因组学研究的最新进展总结如下。
01 第一个茶基因组-云抗10号
云抗10号属于阿萨姆种(CAS),自2010年中科院提出“茶基因组”计划,历经7年,云抗10号基因组得以发表。
文章题目:The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution
of Caffeine Biosynthesis
发表期刊:Molecular Plant
发表时间:2017年6月
组装技术:Illumina(159.43×)
主要结果:茶是世界上最古老、最受欢迎的三大饮料之一,具有重要的经济、医学和文化价值。利用高深度的二代测序,研究者构建了第一个茶树基因组,其组装的基因组大小为3.02G,Contig N50=20 Kb。通过注释,共获得36951个蛋白编码基因,重复序列占比80.89%,由此证明茶树巨大的基因组是由LTR转座子缓慢稳定而长期地扩增引起的。比较基因组分析表明,与类黄酮代谢物合成相关的基因得到显著扩增,这增强了茶儿素的产生,是茶环境适应性的具体体现。最后,研究者选取25种山茶属植物进行比较基因组、转录组分析,表明茶组植物富含茶多酚和咖啡因,这是茶树叶片适合制茶的关键所在;茶树中的咖啡因可能起源于可可,但随后经历了独自的进化。总之,本研究揭示了决定茶叶风味和品质以及生态适应性的遗传基础。

图2 与茶加工、品质相关的三种重要代谢途径(黄酮、茶氨酸、咖啡因)的进化差异
02 中国种(CSS)-舒茶早基因组
作为小叶茶中的明星品种,舒茶早基因组引起科研人员的极大兴趣,先后历经四个版本,多组学的联合应用也充分解析了其与大叶茶的遗传差异、起源进化等。
(1)舒茶早草图-PNAS、Scientific Data
文章题目:Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality
发表时间:2018年3月
组装技术:Illumina(436×)+PacBio(40×)+BAC+RAD-seq
主要结果:茶主要分为两类,中国种和阿萨姆种。第一个发表的茶基因组属于阿萨姆种,中国种尚未有参考基因组。基于此,研究者通过高深度的二代、低深度的三代对明星种舒茶早进行测序组装,组装的基因组大小为3.14Gb,Contig N50=67.07 Kb;通过注释,发现重复序列占比64%,共获得33932个蛋白编码基因。
比较基因组发现,茶树与猕猴桃亲缘关系最近,在8000万年前产生分化,随后茶在38-154万年前进一步分化形成中国种和阿萨姆种;茶树基因组发生过两次WGD事件,导致了与儿茶素类、咖啡碱生物合成相关的基因拷贝数显著增加;与萜烯类合成相关的基因在舒茶早中也显著扩张,解释了茶香的来源。最后结合转录组与转基因分析,研究者证明了CsTSI是茶氨酸合成的关键酶基因。总之,本研究为茶叶风味物质形成机理与品质调控研究提供了新见解。
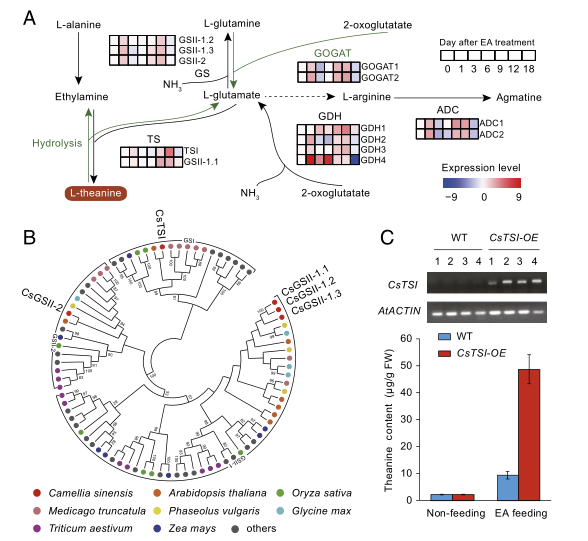
图3 茶氨酸合成途径的解析
值得注意的是,在18年将此篇文章发表在PNAS后,研究者19年7月对前述文章的预测基因进行了重新注释(从33932-变为53512),相关成果以“the tea plant reference genome and improved gene annotation using long-read and paired-end sequencing data”更新在Scientific Data上。
(2)舒茶早精细图-Molecular Plant
文章题目:The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation of tea plants
发表时间:2020年4月
组装技术:Illumina+PacBio(87.2×)+ Hi-C
主要结果:已有的茶树参考基因组仍存在诸多局限性,且已有关于栽培茶树起源进化的研究多是基于质体基因组和简化基因组。基于此,研究者通过基因组+重测序构建了染色体级别的茶树基因组,解析了栽培茶树的起源进化。结合PacBio+Hi-C构建染色体水平的茶树参考基因组(基因组大小为2.94Gb,Contig N50=600.46 Kb),研究者先通过比较基因组发现了茶树基因组中显著扩张并形成基因簇的基因,功能注释表明这些基因显著富集在于茶香气、抗逆性及萜烯生物合成的通路上;随后基于重测序研究,表明现有茶树可分为野生型、阿萨姆型和中国栽培种3类,并结合进化分析表明国内栽培茶树起源于中国西南地区。
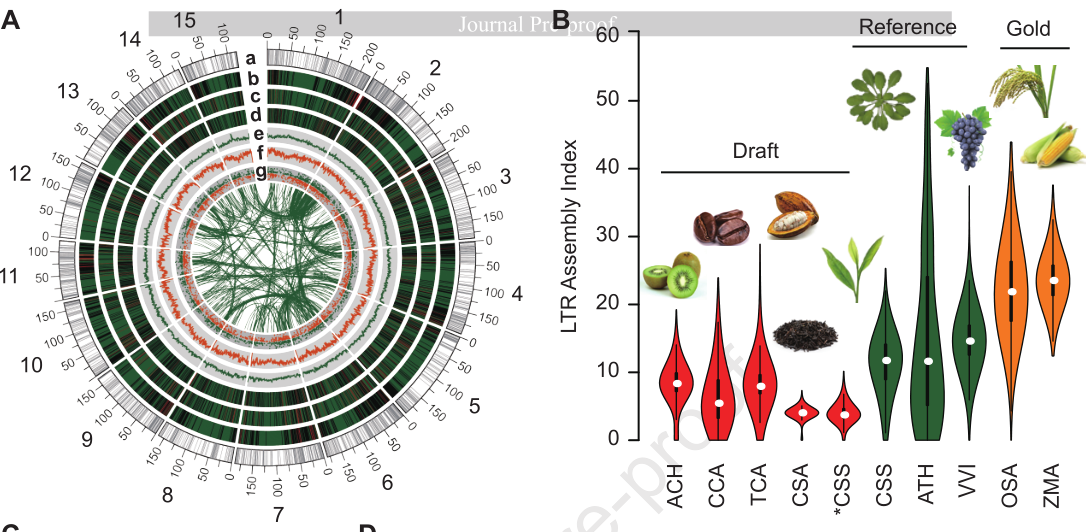
图4 舒茶早基因组及其组装评估
(3)最完整的舒茶早基因组-Horticulture Research
文章题目:The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant
发表时间:2020年5月
组装技术:Illumina+PacBio+Hi-C(113×)
主要结果:此篇文章在18年PNAS文章的基础上,进行了高深度的Hi-C测序,成功构建了Scaffold N50=218.1Mb的染色体级别舒茶早基因组。随后通过比较基因组确定茶树只发生过一次WGD事件,而不是之前报道的两次,且WGD事件导致了串联重复,增加了功能差异基因,在茶特异性生物合成或应激反应中发挥重要作用。最后通过QTL分析确定了64个与儿茶素和咖啡因形成相关的基因,且其中两个来源于WGD事件,这说明WGD事件在茶树资源的多样性和进化中发挥重要作用。
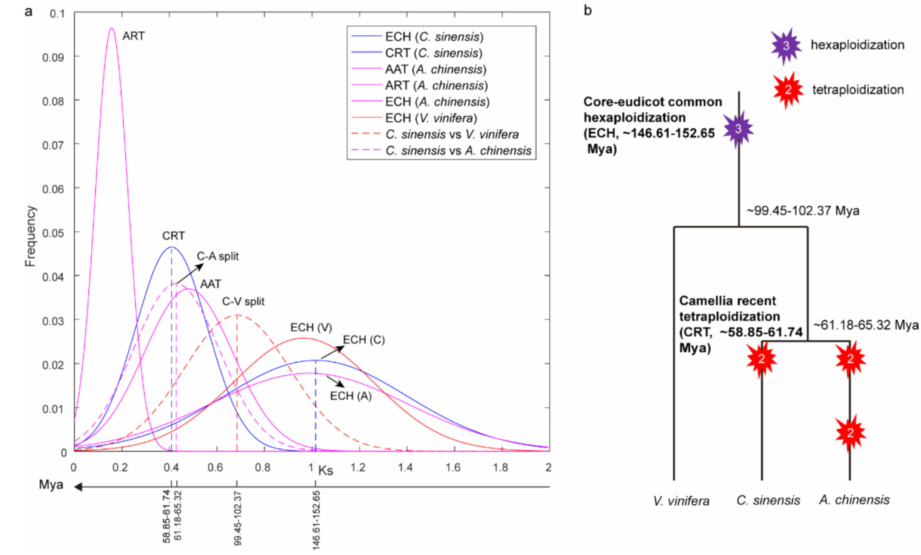
图5 茶树WGD事件的确定
03 其它中国种茶基因组(CCS)-碧云和龙井43
除舒茶早外,制作六大名茶的著名茶品种还包括很多,例如“碧云”和“龙井43”,2020年这两个茶品种也构建了高质量的参考基因组。
(1)碧云基因组
文章题目:The Chromosome-Level Reference Genome of Tea Tree Unveils Recent Bursts of Non-autonomous LTR Retrotransposons to Drive Genome Size Evolution
发表期刊:Molecular Plant 发表时间:2020年4月
组装技术:Illumina+PacBio(127.66×)+Hi-C
主要结果:通过Illumina结合PacBio和Hi-C对‘碧云’茶树品种进行基因组测序,组装的茶树基因组大小为2.92Gb,Contig N50=625.11kb,并将97.87%的序列锚定在15条假染色体上。通过注释,预测到40812个蛋白编码基因,其中85.08%的基因可得到功能注释,研究者认为LTR逆转座子是驱动茶树基因组扩张的首要动力。
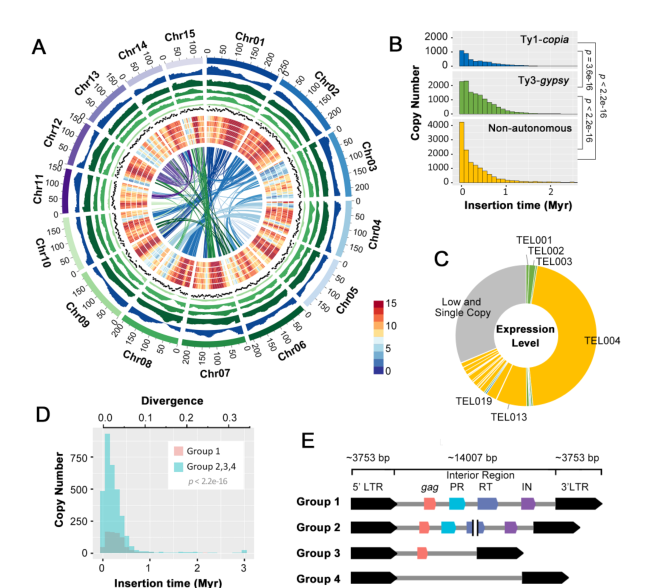
图6 碧云基因组特征
(2)龙井43基因组
文章题目:Population sequencing enhances understanding of tea plant evolution
发表期刊:Nature Communications
发表时间:2020年9月
组装技术:Illumina、PacBio(59×)、Hi-C、10×Genomics、Bionano
主要结果:研究者使用Illumina,PacBio,Hi-C技术构建了LJ43的参考基因组,其基因组大小为3.26Gb,contig N50=271.33Kb,并将70.9%的contig锚定到15条染色体上。随后,研究者通过10×Genomics、Bionano、遗传图谱等技术验证了组装结果的准确性。通过注释,共获得33556个蛋白编码基因,重复序列占比80.06%。WGD分析表明SCZ、LJ43和YK10在25个百万年前发生了相同的WGD事件,且LJ43和SCZ的分化时间早于LJ43和YK10。
随后研究者选取139份材料进行重测序来揭示茶树的起源进化,139份材料可分为三组,分别对应于CSR、CSS和CSA群体。CSR的杂合度显著高于CSA和CSS;LD衰减图表明,CSS的衰减速度慢于CSA。茶树近缘种群是茶变种和阿萨姆变种的祖先,驯化过程中二者的选择方向存在差异,茶变种在驯化过程中与风味相关的萜烯类代谢基因和抗病相关基因受到选择强于阿萨姆变种茶树。
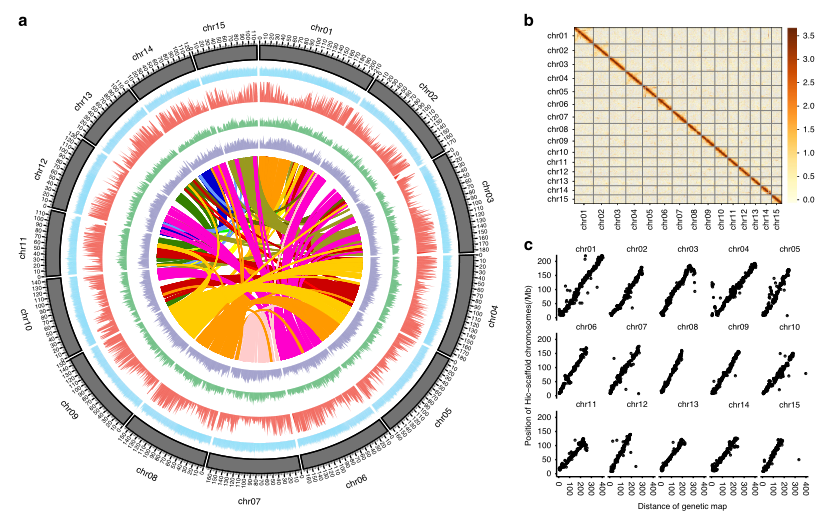
图7 龙井43基因组
04 古茶树基因组
古茶树不仅具有独特的文化与经济价值,而且还是研究茶树起源、分类与品种选育的重要资源。2020年7月,华中农大研究人员以Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties为题,在Nature Communications上发表了高质量的古茶树基因组。
通过PacBio和Hi-C测序,研究者构建的茶树参考基因组大小为3.11G,contig N50=2.6 Mb,并将99.55%的序列挂载到15条染色体上。研究者发现舒茶早茶树基因组相比于本研究组装的古茶树DASZ基因组少了10个R基因,可能与参与生物胁迫反应有关。随后基于转录组和表型数据,将217份茶树材料分为5个亚群,每个亚群的LD衰减均非常显著,说明茶树资源群体具有丰富的遗传多样性。通过GWAS分析,获得176个与儿茶素相关的变异位点;选择性消除分析表明茶树风味并未受到长期的人工定向选择。总之本研究为茶树功能基因组学、分子育种提供新见解。

图8 与茶儿素合成相关的候选基因
总结
通过对上述8篇文章进行总结,我们可看出PacBio+Hi-C是组装茶树基因组的标配技术;基因组+重测序是解析栽培茶树起源进化的必要组学;转录组+代谢组是解析茶风味和口感差异的重要手段;GWAS+QTL+转录组是定位与茶驯化性状相关基因的基础方法。但最重要的,关于栽培茶树的起源进化、WGD事件对茶基因组的影响,尚未完全搞清楚。
展望未来,笔者认为茶树基因组的发展趋势将集中在以下三点:
(1)整合各类型的茶树基因组,构建茶树的泛基因组,来完整、系统的揭示各类茶基因组的遗传差异(需注意HiFi技术的运用)。
(2)选取范围更广、更具代表性的茶树品种(尤其注重加入古茶树)来厘清茶树究竟是起源我国西南地区还是华南地区。
(3)整合多组学,挖掘与茶氨酸、儿茶素、咖啡因相关的基因、转录因子、三维结构差异等,以促进茶树新品种的选育。
诗酒趁年华,期待承载文人骚客精神的茶文化,在基因组学的助力下,继续发扬光大!
 微信公众号
微信公众号


 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|
