
01
菲沙与三代人全基因组重测序技术
永利集团3044三代人类基因组重测序技术基于PacBio seq II e平台和PacBio Revio平台进行三代人全基因组重测序。利用其长读长、无GC偏好性、覆盖度均一等优势可以轻松获取基因组高GC和重复序列区域的信息,那么全新引进的PacBio Revio平台在PacBio seq II e平台优势的基础之上,还拥有超低成本、超级芯片和超高性能,Revio平台每年可完成1300个人类基因组测序,成本低于1000美元,推动HiFi基因组测序进入千元时代。我们非常期待与您合作,共同为人类医学研究做出更多的贡献!

生信分析流程
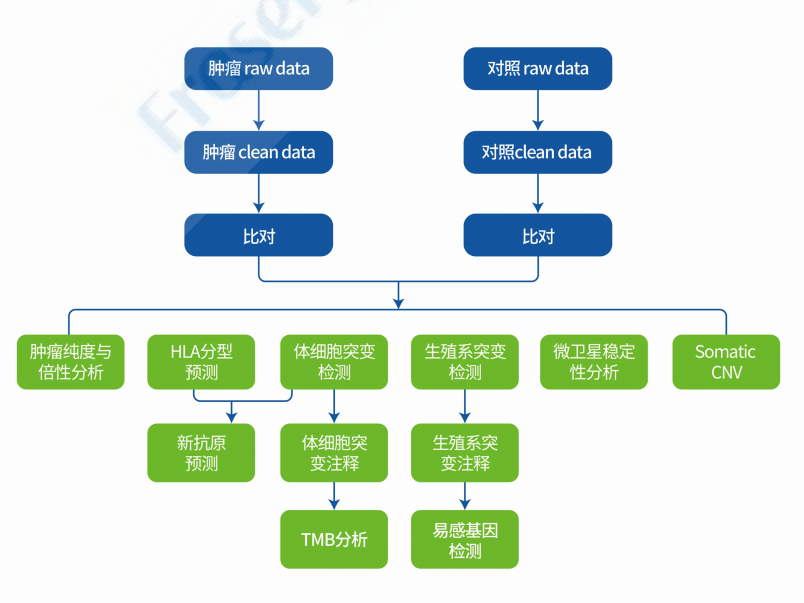
分析内容
1、SNP与InDel检测
SNP(Single Nucleotide Polymorphisms)是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,主要包括转换、颠换,它是人类可遗传的变异中最常见的一种,占所有已知多态性的 90% 以上。SNP 在人类基因组中广泛存在,人体许多表型差异、对药物或疾病的易感性等等都可能与SNP有关。
InDel (insertion-deletion) 是指基因组上发生的小片段(通常定义为1-50bp)的插入和缺失。和SNP一样,大部分的InDel也是核苷酸多态性的表现形式之一。

图1 SNP类型统计柱状图
2、肿瘤纯度与倍性分析
当DNA是从癌细胞和正常细胞的混合组织中取和测序的时候,癌细胞的比例和癌细胞中的基因组的拷贝数信息是不能直接得到的。ABSOLUTE软件根据肿瘤样本基因组的拷贝数和体细胞突变点的等位基因频率,可以计算出肿瘤样本的纯度(Purity)和倍性(Ploidy)。
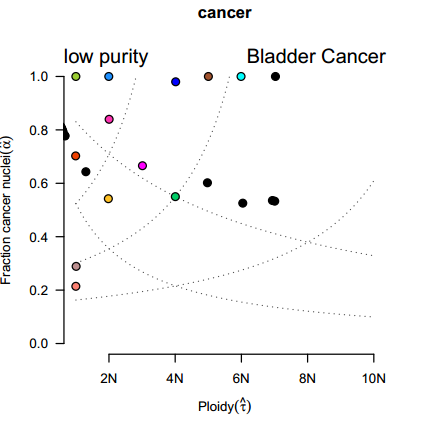
图2 肿瘤纯度和倍性的散点图
3、HLA分型检测
HLA分型(human leucocyte antigen,即人类白细胞抗原)是一个由一系列紧密连锁的基因座位所组成的具有高度多态性的复合体。定位于第6号染色体短臂6P21.31区,长3600kb。是目前所知人体最复杂的多态性系统,每一个基因都具有数千个等位基因。 HLA 是人类组织相容性复合体(MHC)的表达产物,是构成移植排斥反应的重要抗原物质。 HLA 按其分布和功能分为Ⅰ类抗原、Ⅱ类抗原和Ⅲ类抗原。经典的 HLAⅠ类抗原包括 HLA-A、 HLA-B、 HLA-C;HLAⅡ类抗原包括 HLA-DP、 HLA-DQ、 HLA-DR。 CD8 阳性 T细胞能识别癌细胞表面 HLA Ⅰ类分子并破坏癌细胞,并且人体的大部分细胞含有两套编码 HLA Ⅰ类分子的基因:一套基因遗传自母亲,另一套基因遗传自父亲。

表1 HLA分型检测结果
4、TMB分析
肿瘤突变负荷(TMB)被定义为每百万碱基中被检测出的,体细胞基因编码错误、碱基替换、基因插入或缺失错误的总数。肿瘤突变负荷(TMB)作为PD-1 抗体治疗效果评估的最新标志物,其效果已在PD-1抗体用于治疗有错配修复缺陷的结直肠癌中得到证实。
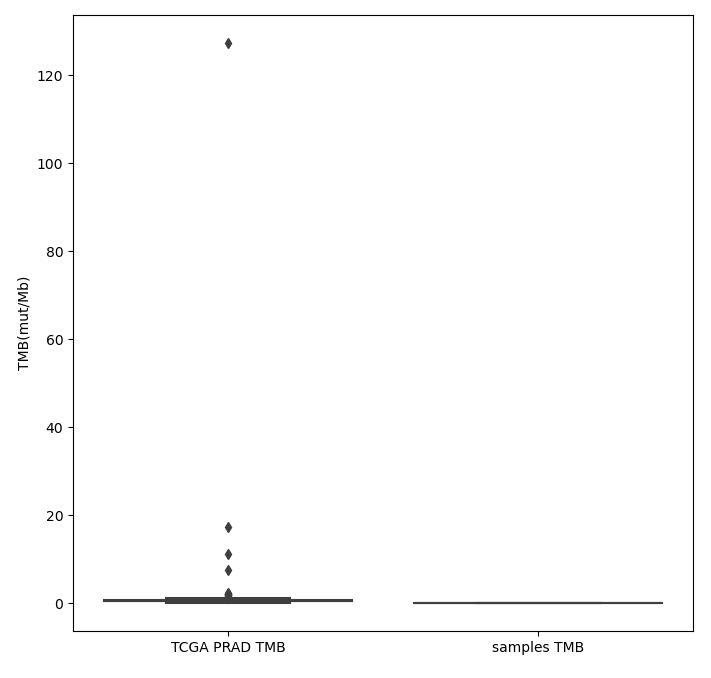
图3 TMB柱状图
02
文献案例
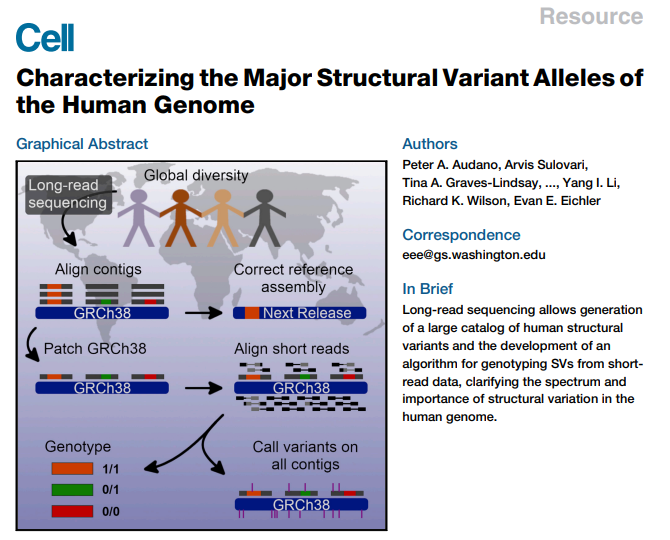
文章题目:Characterizing the Major Structural Variant Alleles of the Human Genome
题目翻译:人类基因组中主要结构变异等位基因的特征分析
发表期刊:Cell(IF:36.216)
材料选择:
11个人类基因组 (包括3个非洲人、2个亚洲人、2个欧洲人、3个美洲人和1个南亚人:2个之前已由本研究测序过的葡萄胎数据 (CHM1和CHM13);2个最近在线的亚洲人基因组AK1和HX1
测序策略:
三代WGS;PacBioRSII、PacBioSequel测序平台,50x以上测序深度
文章摘要:
为了提供一个全面的人类结构变异(SVs)资源,作者生成了长序列数据并分析了15个人类基因组的SVs。作者测序解决了99,604个插入、删除和反转,其中包括2,238个(1.6 Mbp)在所有发现基因组中共享,另外的13053 (6.9 Mbp)存在于大多数基因组中,这表明参考文献中存在较小的等位基因或错误。另外440个基因组的基因分型证实,在独特的常染色质中最常见的sv现在已被序列解析。我们报告了人类染色体最后5mbp的9倍SV偏倚,其中近55%的VNTRs(可变数量)串联重复序列)映射到基因组的这一部分。我们确定了影响编码和非编码调控位点的SVs,从而改进了注释和功能变异的解释。这些数据为构建规范的人类参考提供了框架,并为开发能够捕获等位基因多样性的高级表示提供了资源。
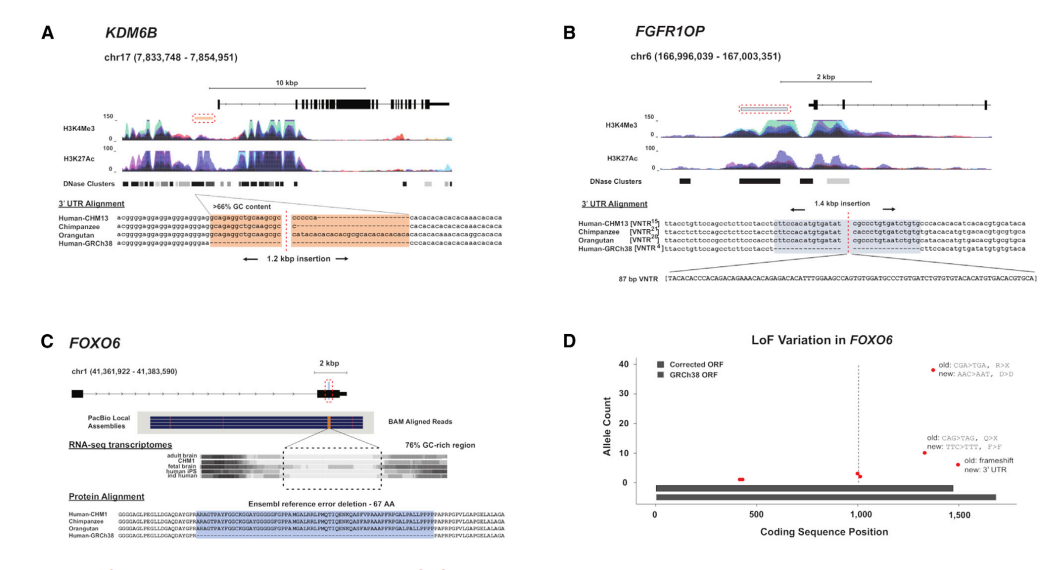
图4 纠正调控元件和FOXO6阅读框架
参考文献:
Audano P A, Sulovari A, Graves-Lindsay T A, et al. Characterizing the major structural variant alleles of the human genome[J]. Cell, 2019, 176(3): 663-675. e19.
 微信公众号
微信公众号


 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|
