
中草药是中华民族的瑰宝,更是我国宝贵的历史文化遗产。神农尝百草、张仲景《伤寒杂病论》、李时珍《本草纲目》,中草药的研究在我国绵延数千年。而近年来,随着屠呦呦教授因青蒿素获诺贝尔奖,有关中草药的研究更是引起国内外研究学者的广泛重视。要全面了解中草药的药用原理与适应机制,基因组测序研究必不可少。
自2014年起,三代长读长测序先后助力铁皮石斛、冬虫夏草、青蒿等20多种中草药高质量参考基因组的发表(表1),为中草药的品质改良和工业化生产提供了理论基础。在此背景下,小编结合4篇中草药基因组高分文章,来浅谈三代测序下中草药基因组研究的新思路。
表1 已发表的中草药基因组(基于三代测序)
1、三代测序助力解析中草药药用成分的合成通路
黄岑基因组
发表概况:2019年4月,Molecular Plant(IF=10.812),The Reference Genome Sequence of Scutellaria baicalensis Provides Insights into the Evolution of Wogonin Biosynthesis
测序技术:Illumina(116X)+PacBio(117X)+10X Genomics(485X)+Hi-C(140X)用于基因组组装;RNA-seq用于转录组分析
研究亮点:汉黄芩素合成通路的解析
研究方案:

图1 黄岑基因组研究技术路线
研究结果:
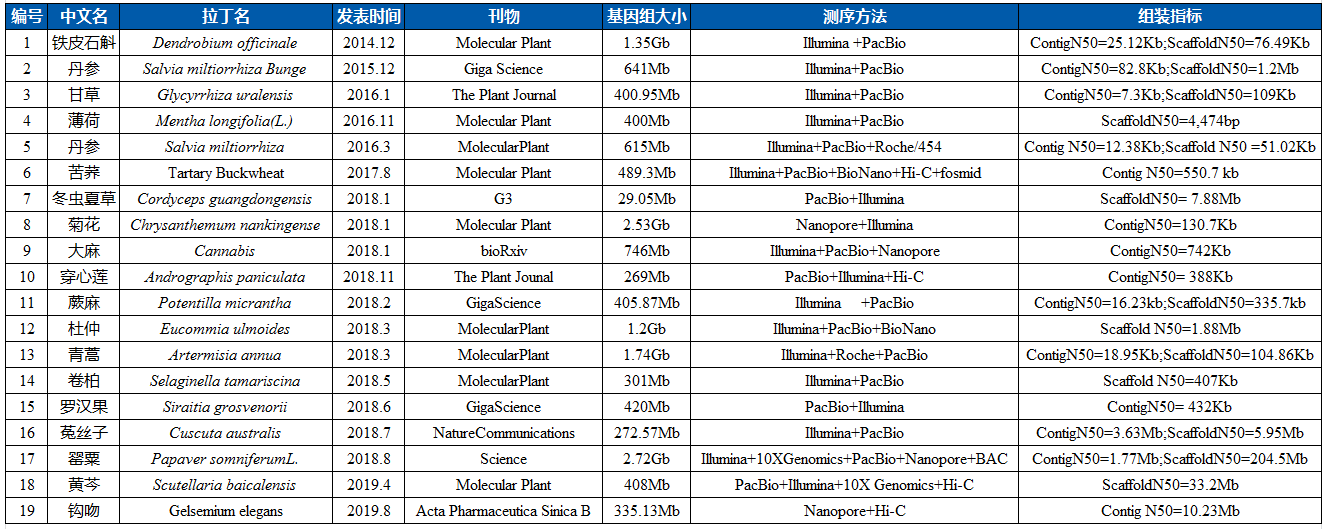
运用PacBio数据组装、Illumina数据纠错、10 X genomics数据延伸片段长度、Hi-C数据挂载染色体,研究者最终构建黄岑的参考基因组大小为386.63 Mb,Scaffold N50=33.2 Mb;通过注释,得到28930个基因,重复序列占比超55.15%,且LTR的含量最丰富。前期研究表明,4’-去氧黄酮类物质仅在黄岑物种中含有,研究者对黄岑及其近缘种(芝麻、丹参)进行基因家族扩张分析,表明黄酮途径合成酶基因只在黄岑中得到扩增,且在物种分化后经串联重复产生。最后,结合酶活和RNAi实验,研究者找到了参与合成黄芩素的酶PFOMT5,从而解析了汉黄芩素的合成途径。总之,该研究为汉黄岑素的人工合成提供了理论基础,同时也为唇形科其他物种的遗传分化提供了参考。
图2 汉黄芩素生物合成候选PFOMT基因的筛选
罂粟基因组
发表概况:2018年10月,Science(IF=41.037),The opium poppy genome and morphinan production
测序技术:Illumina(214 X)+10X Genomics(40X)+PacBio(66.8 X)+ Nanopore+BAC用于基因组组装;RNA-seq用于转录组分析
研究亮点:基因组结构变异;超级基因簇促进止咳那可丁和镇痛吗啡类生物碱合成
研究方案:
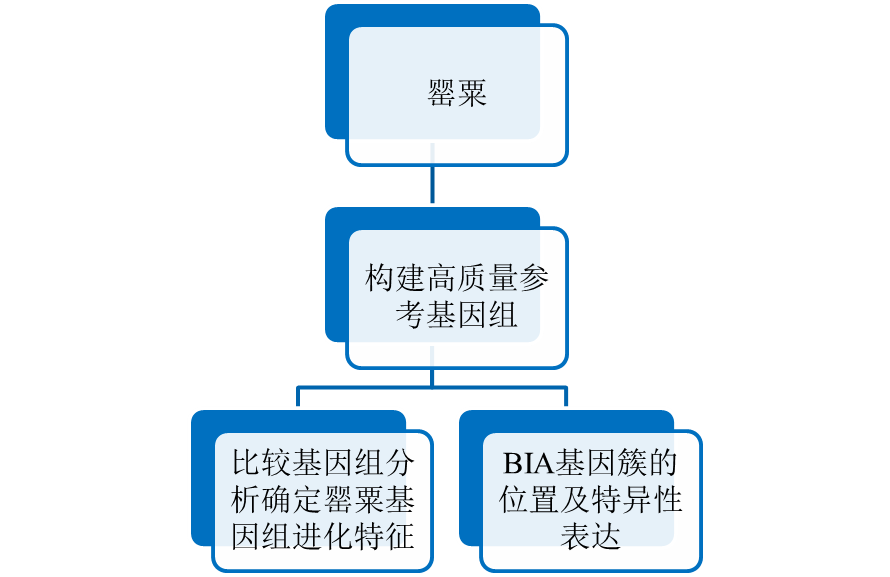
图3 罂粟基因组研究技术路线
研究结果:
罂粟基因组重复序列占比高(超70%),且经历多次大规模结构变异,在此背景下,结合多种测序技术,研究者构建了罂粟参考基因组(基因组大小2.71Gb,Contig N50=1.77 Mb)。通过比较基因组分析,发现罂粟在780万年前出现一次全基因组加倍事件;在毛茛科和罂粟科产生分化之前(大约距今1.1亿年)发生一次基因组部分片段加倍。随后,研究者在11号染色体上鉴定出15个基因形成的超级基因组(BIA),BIA与那可丁和吗啡类生物碱的生物合成有关(形成STORR基因)。此外,罂粟基因组经历的多次结构变异(加倍、扩增、丢失、融合和重排)等促进了BIA的形成,且BIA在罂粟的根、茎部特异表达且共表达。该研究为罂粟为药用价值开发、进化历史探究奠定重要基础。
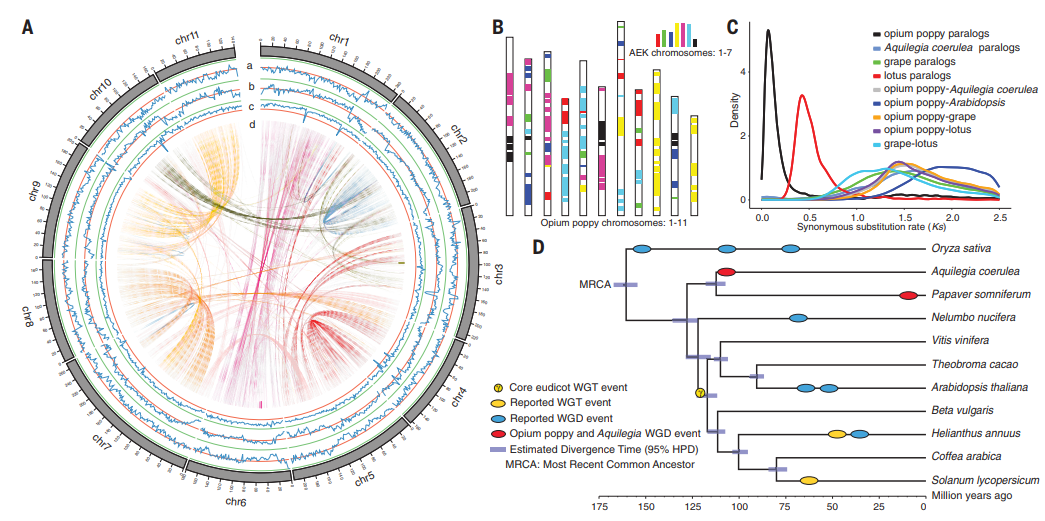
图4 罂粟基因组的进化特征
小结
科学研究表明,中草药的药用成分大多是其次生代谢物,因此基于三代测序解析中草药的次生代谢物的合成尤为重要。其研究思路可概括如下:
对于特有次生代谢产物,通过基因家族扩张与收缩分析确定物种特异性基因家族,随后采用转录组进行该代谢通路关键基因表达模式的分析,最终解决其合成通路的解析。
2、三代测序助力解析中草药的适应机制
菟丝子基因组
发表概况:2018年7月,Nature Communications (IF=11.878),Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis
测序技术:Illumina(116X)+PacBio(97.6X)用于基因组组装;RNA-seq用于转录组分析
研究亮点:菟丝子寄生机制的解析
研究方案:
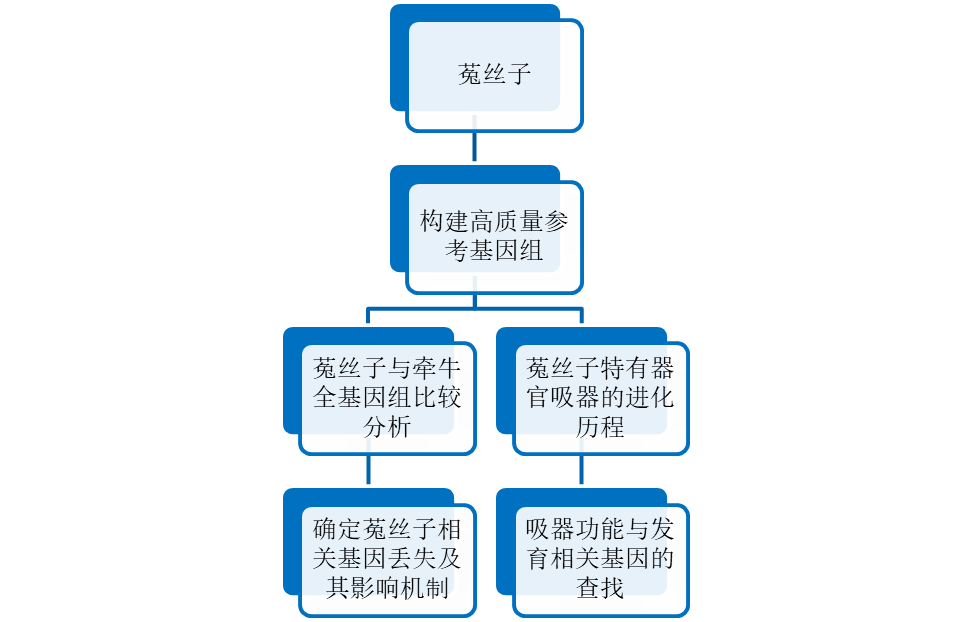
图5 菟丝子基因组研究技术路线
研究结果:
利用三代数据组装和二代数据纠错,研究者构建的菟丝子基因组大小为272.57 Mb,ContigN50=3.63Mb,ScaffoldN50=5.95Mb。随后,研究者将菟丝子基因组与牵牛基因组进行系统进化树分析和共线性分析,发现两者的共同祖先在750万年前经历了一次全基因组三倍化加倍事件。在此之后,牵牛保持加倍事件后的基因(蛋白编码基因42783),而菟丝子则经历了剧烈的基因丢失事件(蛋白编码基因19671),且其丢失的基因主要与光合作用、根和叶的功能与发育、抗性基因的转录调控等相关,从基因组层面解释了菟丝子寄生生活的适应机制。最后,结合转录组数据、基因家族扩张分析,确立了诸多与吸器功能与发育相关的基因。总之,该研究为菟丝子属植物的演化及寄生植物与寄主间互作关系的研究提供重要新见解。

图6 菟丝子相关基因的丢失
铁皮石斛基因组
发表概况:2015年,Molecular Plant(IF=10.812),The Genome of Dendrobium officinale Illuminates the Biology of the Important Traditional Chinese Orchid Herb
测序技术:Illumina(133X)+PacBio(10X)
研究亮点:铁皮石斛适应机制的解析(抗旱、共生等)
研究方案:
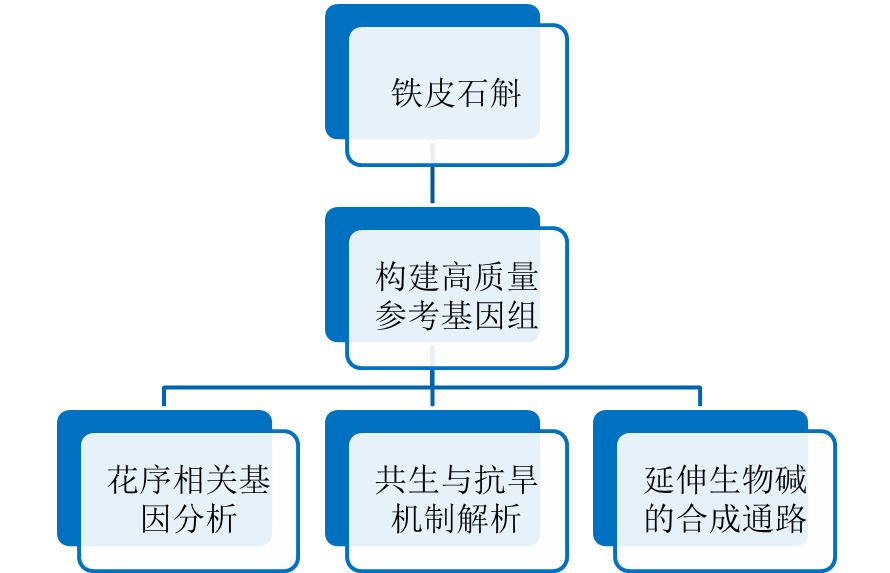
图7 铁皮石斛基因组研究技术路线
研究结果:
研究者通过二代和三代测序,构建的铁皮石斛基因组大小为1.35 Gb,ContigN50=25.12 kb。基于高质量参考基因组,研究者进一步进行比较基因组学分析,结果表明兰科植物有着完整的花序基因集,且相对于单子叶植物具有特异性的花序基因;与真菌共生和抗旱性有关的一些基因家族发生了明显扩增;解析铁皮石斛药用成分的生物合成通路,发现与多糖合成相关的SPS和SuSy基因发生大规模复制,从而可以将铁皮石斛生物碱的合成通路延伸到16-epivellosimine步骤。总之,本研究为铁皮石斛共生机制的解析、药用成分的合成与利用奠定重要基础。
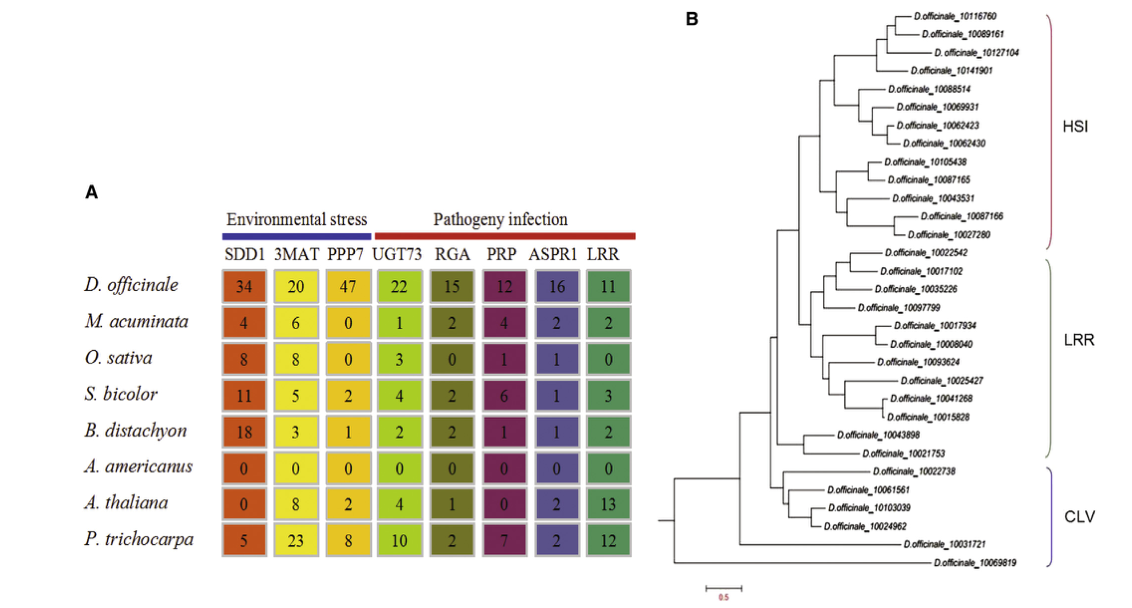
图8 铁皮石斛扩张基因分析
小结
如其他植物一样,许多中草药在进化历程中表现出独特的生命活动,而通过基因家族收缩扩张,同时结合转录组数据,可从基因组层面解析中草药的适应机制。概括来讲,三代测序下关于中草药适应机制的研究思路主要有如下几点:
(1)解析共生/寄生机制,一方面是解析中草药与真菌的共生机制;另一方面是解析某些中草药的寄生机制。
(2)解析进化历程,通过比较基因组将研究物种与近缘物种进行共线性分析,可以解析中草药的进化历程与进化地位。
(3)挖掘抗性基因,通过基因家族收缩扩张分析,解析某些中草药独特的抗性机制(如牛耳草抗旱等),然后结合转录组数据,鉴定不同条件下的差异表达基因,最后再结合相关生理实验进行验证。
总之,在三代测序下,中草药基因组研究将会达到新的高度。
参考文献:
[1] Zhao Q, Yang J, Cui M Y, et al. The Reference Genome Sequence of Scutellaria baicalensis Provides Insights into the Evolution of Wogonin Biosynthesis[J]. Molecular plant, 2019.
[2] Guo L, Winzer T, Yang X, et al. The opium poppy genome and morphinan production[J]. Science, 2018, 362(6412): 343-347.
[3] Sun G, Xu Y, Liu H, et al. Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis[J]. Nature communications, 2018, 9(1): 2683.
[4] Yan L, Wang X, Liu H, et al. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb[J]. Molecular plant, 2015, 8(6): 922-934.
 微信公众号
微信公众号


 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|
